Specifying additional/alternative basis functions
Gaussian has an extensive collection of built-in options for basis sets. The included basis sets will be sufficient for the vast majority of computational needs; however, the need may arise for additional basis functions or further customization, which can be accomplished using the gen and extraBasis keywords.
gen
gen allows you to specify your own basis set specifications to the atom level. It is used in place of a basis set keyword in the routing line (e.g. #N M062X/gen). The basis set specification is then read after the molecule specification. The basis definition input can either be entered directly into the input file or read into the input stream using g16’s include (@) function as described earlier in the course.
The basis specification can either utilize g16’s built-in basis functions or custom-defined basis definitions, but they must follow a specific format:
#N M062X/gen ...
...
{Molecule geometry}
<<Blank line>>
H 0 ! {atomSymbol} 0
S 3 1.00 ! these are the basis functions
0.122518D+02 0.233371D-01 ! from Jensen's pcseg-1
0.186871D+01 0.159150D+00
0.418208D+00 0.500000D+00
S 1 1.00
0.106100D+00 1.0000000
P 1 1.00
0.100000D+01 1.0000000
**** ! **** tells g16 that its done
He 0 ! reading functions for this atom
S 3 1.00 ! and to move to the next
0.368654D+02 0.273556D-01
0.558021D+01 0.174855D+00
0.119170D+01 0.500000D+00
S 1 1.00
0.268925D+00 1.0000000
P 1 1.00
0.145000D+01 1.0000000
****
<<More atoms>> ! add as many as you need
**** ! this section ends with ****
<<Blank line>> ! the g16 input file ends with
<<Blank line>> ! 2 blank lines
If Gaussian encounters an atom that doesn’t appear in the molecule it will kill the run (this is a safety feature meant to detect input mistakes). This can be circumvented by reformatting the atom definition line from
{atomSymbol} 0to-{atomSymbol}, (in fact, this is how g16’s own built-in basis sets are defined). This is useful when creating standard basis set files for amy atoms which are intended to be read directly into the input stream using the include (@) function.
By default specifying a basis set in the routing line applies it to all atoms in the calculation. This is usually appropriate (given you’re using an advanced enough basis set) however, it may sometimes be necessary to “mix-and-match” basis sets (when calculating transition metal complexes using the Pople basis family, for example). We can also do this using the gen keyword with the following format.
Say we want to optimize a cisplatnin3 molecule using the 6-31G(d,p) basis set. We’re going to run into an issue since 6-31G(d,p) isn’t defined for atoms larger than krypton. We’ll have to select a different basis set to use for platinum; a common choice of basis sets for transition metals is LANL2DZ, however the non-ECP part of the LANL2DZ definitions is small which has raised concerns on its accuracy. I recommend using the Karlsruhe basis sets (def2-XZVP) which are defined for atoms up to radon.
#N M062X/gen ...
...
{Molecule geometry}
<<Blank line>>
H N Cl 0 ! for all Cl, N, & H
6-31G(d,p) ! use 6-31G(d,p) basis functions
****
Pt 0 ! for all Pt
LANL2DZ ! use LANL2DZ basis functions
**** ! section ends with ****
<<Blank line>> ! the g16 input file ends with
<<Blank line>> ! 2 blank lines
Now, say for some reason we wanted to use a different basis set for just one of the chlorine atoms (you’ll see why we might want to do that in the exercise at the end of this lesson). gen allows us to do that too, as long as we know the number of the atom we want to modify (we can get this from a modeling program like GaussView or Avogadro). In this example, we’ll use the 6-31+G(d,p) basis set to describe atom #3 (one of the chlorines) and 6-31G(d,p) to describe atom #2 (the other chlorine).
#N M062X/gen ...
...
{Molecule geometry}
<<Blank line>>
H N 2 0 ! for all N, H & atom #2 (Cl)
6-31G(d,p) ! use 6-31G(d,p) basis functions
****
3 0 ! for atom #3 (Cl)
6-31+G(d,p) ! use 6-31+G(d,p) basis functions
****
Pt 0 ! for all Pt
LANL2DZ ! use LANL2DZ basis functions
**** ! section ends with ****
<<Blank line>> ! the g16 input file ends with
<<Blank line>> ! 2 blank lines
extraBasis
The extraBasis keyword works similarly to gen, but with a little reduced functionality. extraBasis works only to add extra basis functions to the calculation, i.e. if your selected basis set has function definitions for an atom already extraBasis will use additional basis functions for that center on top of those already define. extraBasis creates more room for input error, while providing limited functionality over gen.
genis the safer and more functional way to utilize custom basis definitions.
The syntax of extraBasis is identical to that of gen, however, instead of replacing the basis functions for the designated atoms it will add them to the existent ones.
The latter examples of the previous section can also be implemented using extraBasis. Since, no basis functions are defined for platinum in 6-31G(d,p), extraBasis adds the LANL2DZ basis functions to nothing.
#N M062X/6-31G(d,p) extraBasis ...
...
{Molecule geometry}
<<Blank line>>
Pt 0 ! for all Pt
LANL2DZ ! use LANL2DZ basis functions
**** ! section ends with ****
<<Blank line>> ! the g16 input file ends with
<<Blank line>> ! 2 blank lines
To add diffuse functions to just atom #3 (Cl) use extraBasis and specify only the diffuse function. N.b. specifying 6-31+G(d,p) in the extraBasis section would add all of the 6-31+G(d,p) functions to the atom.
#N M062X/6-31G(d,p) extraBasis ...
...
{Molecule geometry}
<<Blank line>>
3 0 ! for atom #3 (Cl)
SP 1 1.00 ! add diffuse function from 6-31+G(d,p)
0.4830000000D-01 0.1000000000D+01 0.1000000000D+01
****
Pt 0 ! for all Pt
LANL2DZ ! use LANL2DZ basis functions
**** ! section ends with ****
<<Blank line>> ! the g16 input file ends with
<<Blank line>> ! 2 blank lines
This method requires identifying the diffuse basis functions using the differences between diffusion- and non-diffusion-augmented basis set definitions using from the basis set exchange. While this method may work for one or two atoms it does not scale well. So, if you find yourself in the position of needing to augment your calculations like this, gen will be much easier to use on scale.
Effective Core Potentials (a.k.a pseudopotentials)
The computational cost of a calculation scales with the size of the system, namely the number of electrons. As the number of electrons in your system increases, the longer a calculation will take. Typically this isn’t an issue, however optimizations of large systems, for example, organometallic systems with multiple metal centers and complex ligands, can quite quickly become unwieldy. The use of effective core potentials (ECPs or pseudopotentials) is a method for combatting resource and time intensive calculations.
An effective core potential is a basis function (a pseudo-orbital) that is used to “substitute” the inner (core) electrons of an atom. The pseudo-orbitals are formulated to be nodeless in the core region (Figure 1).4 It provides a relativistic effective potential for each core orbital eliminating the need for core basis functions and leading to a substantial reduction in computational effort. Most ECPs are built in a form defined by Pacios and Christiansen (1985).5 A more formal explanation can be found in ref 4.
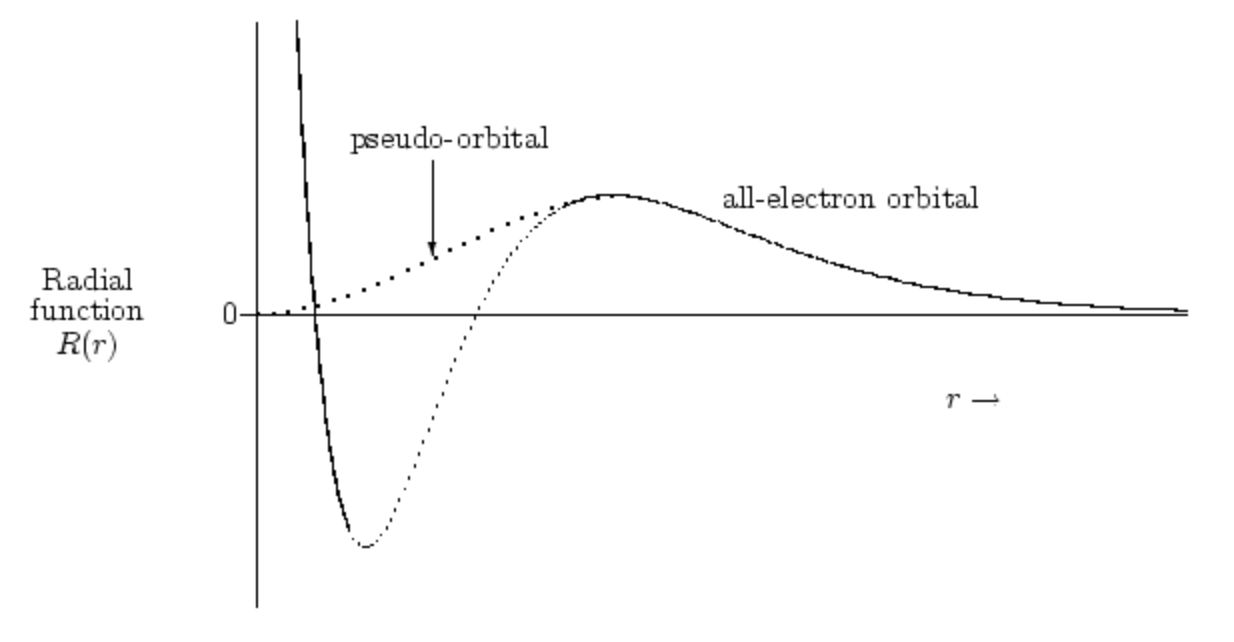 Figure 1. Typical shape of pseudo-orbital. Reproduced from ref 4.
Figure 1. Typical shape of pseudo-orbital. Reproduced from ref 4.
One of the largest questions in the design of ECPs is what constitutes a “core” electron. ECPs come in two “flavors:” large-core and small-core. Large-core ECPs typically substitute all but the outer shell electrons while small-core ECPs substitute all but the outer two shells. For example, consider the three representations of bromine (Figure 2).6 Of the two, small-core ECPs are more expensive to use, but typically demonstrate much more acceptable accuracy.7
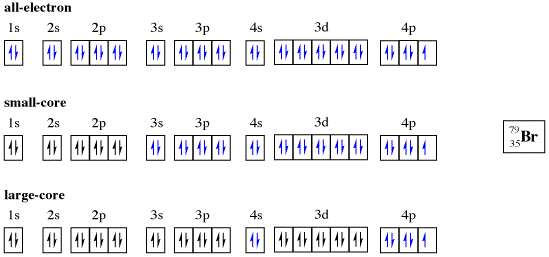 Figure 2. All-electron, small-core, and large-core representations of bromine. Core electrons are designated in black while quantum mechanically treated electrons are denoted in blue. Reproduced from ref 6.
Figure 2. All-electron, small-core, and large-core representations of bromine. Core electrons are designated in black while quantum mechanically treated electrons are denoted in blue. Reproduced from ref 6.
To utilize ECPs in Gaussian use the pseudo=read or genECP keywords.1,8 Their usage is the same, but genECP is equivalent to gen pseudo=read; when adding ECPs to a pre-defined basis set you must use pseudo=read.
#N M062X/def2tzvp genECP ... ! this will not work
#N M062X/def2tzvp pseudo=read ... ! this will work
#N M062X/gen pseudo=read ... ! this will work and is the same as:
#N M062X/genECP ...
Specification of the ECP in Gaussian takes a format similar to gen. It can either be read in using built-in definitions (some basis sets have built in ECPs defined for certain atoms) or defined explicitly.
#N M062X/genECP ...
...
H N Cl 0 ! for all N, H & Cl
6-31G(d,p) ! use 6-31G(d,p) basis functions
****
Pt 0 ! for all Pt
def2tzvp ! use def2tzvp basis functions
****
<<Blank line>> ! blank line starts the ecp block
Pt 0 ! for all Pt
def2tzvp ! use def2tzvp ECPs
<<Blank line>> ! blank line ends the ecp block
...
N.b. when defining multiple ECPs, there is no divider (****) between the atoms, for example:
PD 0
PD-ECP 3 28
f potential
2
2 13.2700000 -31.92955431
2 6.6300000 -5.39821694
s-f potential
4
2 12.4300000 240.22904033
2 6.1707594 35.17194347
2 13.2700000 31.92955431
2 6.6300000 5.39821694
p-f potential
4
2 11.0800000 170.41727605
2 5.8295541 28.47213287
2 13.2700000 31.92955431
2 6.6300000 5.39821694
d-f potential
4
2 9.5100000 69.01384488
2 4.1397811 11.75086158
2 13.2700000 31.92955431
2 6.6300000 5.39821694
PT 0
PT-ECP 3 60
f potential
1
2 3.30956857 24.31437573
s-f potential
3
2 13.42865130 579.22386092
2 6.71432560 29.66949062
2 3.30956857 -24.31437573
p-f potential
3
2 10.36594420 280.86077422
2 5.18297210 26.74538204
2 3.30956857 -24.31437573
d-f potential
3
2 7.60047949 120.39644429
2 3.80023974 15.81092058
2 3.30956857 -24.31437573
Exercise
Once you think you understand everything, take a look at this problem to see if you can compare the isomerization energies of 3-(4-nitrophenyl)but-2-en-2-yl triflate.
References
(1) gen
(2) Unifying General and Segmented Contracted Basis Sets. Segmented Polarization Consistent Basis Sets by Frank Jensen J. Chem. Theory Comput. 2014, 10 (3), 1074
(3) cisplatnin
(4) Effective core potentials by Prof. William Lester (Berkley)
(5) L.F. Pacios and P.A. Christiansen “Ab initio relativistic effective potentials with spin‐orbit operators. I. Li through Ar” J. Chem. Phys. 1985, 82, 2664
(6) Effective Core Potentials (ECP) by Prof. Hendrik Zipse (LMU)
(7) X. Xu and D.G. Truhlar “Accuracy of Effective Core Potentials and Basis Sets for Density Functional Calculations, Including Relativistic Effects, As Illustrated by Calculations on Arsenic Compounds” J. Chem. Theory Comput. 2011, 7 (9), 2766
(8) pseudo